2021年11月20日
梁啟信教授
香港大學李嘉誠醫學院
眼科學系系主任及臨床教授
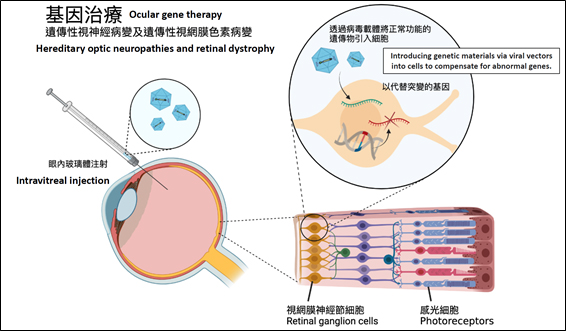
視網膜營養性萎縮(retinal dystrophy)和遺傳性視神經病變(hereditary optic neuropathies)都是罕見的致盲眼疾。前者是感光細胞(photoreceptors)的漸進性功能喪失,而後者則是視網膜神經節細胞(retinal ganglion cells)的漸進性功能喪失。視網膜營養性萎縮最常見的病型是視網膜色素病變,而遺傳性視神經病變的常見病型分別是顯性遺傳視神經病變 (dominant optic atrophy) 和萊伯氏遺傳性視神經病變(Leber hereditary optic neuropathy,簡稱LHON)。
視乎基因突變的種類,部分患者在童年時或已出現嚴重的視力障礙,亦有患者的視力可維持正常逾數十年。這些遺傳性的眼疾一度被視為無法醫治,直至2017年,美國食品及藥物管理局首次批准利用基因治療藥物Luxturna,治療罕見的視網膜營養性萎縮病——萊伯氏先天性黑蒙症(Leber’s congenital amaurosis,簡稱 LCA)。這不單是神經保護治療和基因治療的重大進展,更為遺傳性眼疾的患者,燃點重獲視力的希望。
大多數基因治療的基礎,都是透過病毒載體,將遺傳物質引入細胞以補償異常基因。例如Luxturna提供了正常RPE65基因的功能副本,代替突變的 RPE65 基因,以改善LCA患者的視力。早期臨床數據也表明,基因治療亦可改善LHON患者的視力。近年有不少國際臨床試驗,針對研究眼疾的基因治療和神經保護治療,預計未來可讓更多視網膜營養性萎縮和遺傳性視神經病變的患者受惠。
港大醫學院眼科學系正重點研究關於遺傳性視神經病變和視網膜色素病變的基因治療和神經保護治療。有興趣參與或了解這些研究項目的患者,可以透過電郵(洪小姐:gpchung@hku.hk)或致電 3910 2640 聯絡負責該項目的行政研究協調員。
<刊載於《東方日報》,2021年11月20日>