
17 July 2019
Engineering clinically useful proteins such as antibodies and enzymes requires optimisation of their amino acid sequences to achieve desired functions for therapeutic and research applications. In recent years, CRISPR-Cas enzymes have become important tools in medical research for gene editing. Worldwide efforts towards engineering CRISPR-Cas enzymes have introduced various amino acid changes to improve their editing accuracy and efficiency, and enhance safety and efficacy in correcting gene mutations in patients.
A research team from the School of Biomedical Sciences, LKS Faculty of Medicine, The University of Hong Kong (HKUMed), has developed the first platform for assembling and barcoding protein-encoding sequences carrying multiple mutations en masse and has coupled it with next-generation sequencing to track all variants in an unprecedented throughput. The research has just been published in Nature Methods (link to the publication) and a patent application has been filed based on this work.
Background
The current challenge for engineering CRISPR-Cas enzymes, as well as other proteins, lies in identifying the optimal sets of amino acid changes without under- or over-engineering the protein. The combined effect of multiple amino acid changes is difficult to predict, and the number of possible protein-variant combinations increases dramatically with every additional amino acid being modified. Traditional site-directed mutagenesis techniques for creating protein variants one-by-one are not feasible for generating a large library of variants with multiple amino acid changes.
Finding new ways to enhance the throughput in assembling and accessing the function of protein variants is important for understanding epistatic mutations and accelerating next-generation engineering of genome-editing enzymes with new or improved properties.
The HKUMed research team has developed the first platform for assembling and barcoding protein-encoding sequences carrying multiple mutations en masse and has coupled it with next-generation sequencing to track all variants in an unprecedented throughput.
Pre-existing methods of site-directed mutagenesis and screening are more laborious and time-consuming, and are typically applied in bacterial cells, which limits their utility for engineering genome-editing enzymes and other therapeutic proteins for use in human cells. Using its newly developed platform, named CombiSEAL, the HKUMed team successfully identified new high-fidelity variants of a genome-editing Cas9 protein that have enhanced gene-editing specificity without sacrificing potency and broad targeting range. This high throughput-screening platform aims to lead discovery and optimisation of more clinically useful proteins. The low-cost, easy-to-implement and scalable strategy helps researchers excel in the increasingly competitive environment of innovative bioproducts.
Study results
Streptococcus pyogenes Cas9 (SpCas9) nuclease is the most widely used CRISPR-based genome editing enzyme. Using CombiSEAL, a library of 948 SpCas9 variants, each of which contains one-to-eight mutations, was assembled and delivered into human cells, and their genome-editing efficiency and specificity were systematically quantified. The total number of SpCas9 protein variants characterised in this study greatly exceeds the sum of all other major studies reported thus far (see Table 1 in the PPT).
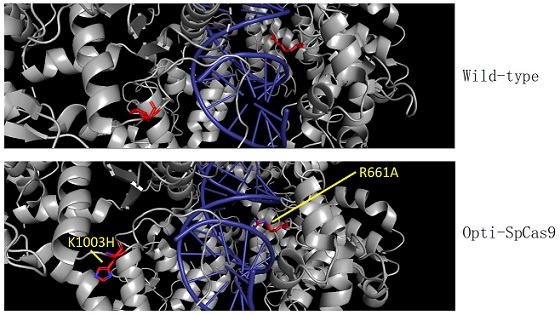
The new HKUMed strategy will allow the team to further easily scale up the engineering process in future. The team also identified new high-fidelity SpCas9 variants, Opti-SpCas9 and OptiHF-SpCas9, which generate much fewer undesired off-target genome edits without sacrificing on-target editing efficiency and broad targeting range. CombiSEAL greatly enhances the throughput for functional impact analysis of a massive number of mutation combinations by a simple one-pot reaction library assembly and a round of short-read sequencing of the variant-specific barcode combination.
Significance of the study
CombiSEAL as developed by the HKUMed team is the first-ever platform that breaks through limits to rapidly and simultaneously profile multiple mutations for protein engineering and map relationships between mutations. This platform can be easily implemented in many laboratories for the massively parallel engineering of proteins relevant to a multitude of biomedical, therapeutic and biotechnology applications. Lead researcher of the study, Dr Alan Wong Siu-lun, Assistant Professor of the School of Biomedical Sciences, HKUMed remarked, “This is a large-scale build-and-test platform for combinatorial optimisation of proteins. The one-pot assembly and barcoding strategy substantially reduces time and cost for the whole process starting from building to functional characterisation of the entire variant library”.
The team identified new variants of SpCas9 with properties that are important for many of the applications enabled by genome editing. Dr Wong stated, “Genome-editing tools need to be accurate to maximise safety for clinical use, while high editing efficiency and a broad target range are also important for applications like CRISPR screens”. Unlike other reported high-fidelity SpCas9 enzymes, Opti-SpCas9 is the only variant compatible with gRNAs containing an additional 5’ guanine for the transcription under the U6 promoter without comprising its on-target activity (see Table 2 in the PPT). This feature is important because U6 is the most widely used promoter in the many CRISPR-based genetic screens performed worldwide.
Previous study results
Dr Alan Wong has been developing various high throughput-screening technologies to systematically dissect the complex genetics underlying diseases including cancers and from there devising new therapeutic strategies. Previous work, published in Nature Biotechnology in 2015 and Proceedings of the National Academy of Sciences in 2016, focused on developing CombiGEM technology to screen genetic interactions. The team also published a review article in the Annual Review of Genetics in 2016 highlighting the potential of that technology in addressing practical challenges in biomedical research. The new CombiSEAL technology presented in this current study extends the scope and impact of combinatorial genetics to engineer proteins for gene-editing applications.
About the research team
This study was led by Dr Alan Wong Siu-lun, Assistant Professor of the School of Biomedical Sciences, HKUMed. Dr Gigi Choi Ching-gee, Post-doctoral Fellow of the School of Biomedical Sciences, HKUMed, was the first author of this work. Dr Peng Zhou, Ms Chaya Yuen Tsz-lo, Ms Becky Chan Ka-ching, Mr Feng Xu, and Ms Dawn Thean (respectively Post-doctoral Fellow, Postgraduate Students, and Research Assistant at the HKUMed School of Biomedical Sciences) were co-authors and they also performed the experiments. Other researchers were Dr Zongli Zheng, Ms Siyu Bao, Ms Athena Chu Hoi-yee from the Ming Wai Lau Centre for Reparative Medicine, Karolinska Institutet in Hong Kong, and Dr Chris Wong Koon-ho and Dr Kaeling Tan from the Faculty of Health Sciences, University of Macau in Macau.
Acknowledgements
This work was supported by funds for Dr Alan Wong Siu-lun’s team including The University of Hong Kong start-up and internal funds, the Croucher Foundation Start-up Allowance and the Hong Kong Research Grants Council (ECS-27105716 and TRS-T12-710/16-R). Other support came from the Swedish Research Council (2016-02830) and the National Natural Science Foundation of China (81672098) (for Dr Zheng Zongli’s team); and from the Science and Technology Development Fund of Macau S.A.R. (FDCT 085/2014/A2), the Research Services and Knowledge Transfer Office of the University of Macau (MYRG2016-00211-FHS and MYRG2018-00017-FHS), and the Start-up fund from the Faculty of Health Sciences, University of Macau (for Dr Chris Wong Koon-ho’s team).
Media enquiries
Please contact LKS Faculty of Medicine of The University of Hong Kong by email (medmedia@hku.hk).

Follow HKUMed