
Professor MH Sham
Dr Vincent CH Lui
In this theme we study molecular signaling in cells and in animal models including mouse, zebrafish and chick. We apply genomic and gene knockout/knockdown technologies to study the changes in cell interactions and signal transduction pathways in a number of developmental processes that have implications in human diseases, such as skeletal development, craniofacial development, haemopoiesis, neural development, neural crest development, cardiovascular and inner ear development and other congenital birth defects.
Animal models of disorders developed by members include cartilage and bone malformations, craniofacial disorders, cardiovascular defects, hearing and balance disorders, neurocristopathy. Several multidisciplinary research programmes operating in the Centre link the two themes Developmental and Functional Genomics and Genetics of Disorders. An example is the AoE programme on "Developmental Genomics and Skeletal Research".(http://web.hku.hk/~aoebioc/) The research team, a model of cooperation between scientists and clinicians from HKU, HKUST, and PolyU employs a variety of state-of-the-art technologies in genomics, proteomics, cell biology and transgenic animal models.
New Deafness and Balance Gene
 Nearly 10 years ago, Prof Kathryn Cheah and her postgraduate students accidentally generated a mouse mutant (Yellow submarine or Ysb) with hearing and balance problems. Through studying Ysb and another similar deaf and balance-impaired mouse mutant, Light coat and circling (Lcc), Prof Cheah and her UK collaborators have discovered that a single factor, SOX2, is required for the development of all the sensory organs of the inner ear. This discovery implicates defects in the regulation of SOX2 gene expression as a cause of human congenital deafness. This finding has important implications for the future development of regenerative therapies in regard to treatment of deafness.
Nearly 10 years ago, Prof Kathryn Cheah and her postgraduate students accidentally generated a mouse mutant (Yellow submarine or Ysb) with hearing and balance problems. Through studying Ysb and another similar deaf and balance-impaired mouse mutant, Light coat and circling (Lcc), Prof Cheah and her UK collaborators have discovered that a single factor, SOX2, is required for the development of all the sensory organs of the inner ear. This discovery implicates defects in the regulation of SOX2 gene expression as a cause of human congenital deafness. This finding has important implications for the future development of regenerative therapies in regard to treatment of deafness.
Aging Gene
 The Hutchison-Gilford Progeria Syndrome (HGPS) is a disease that causes the onset of severe premature aging. HGPS patients have elderly-like appearance and rapid deterioration of physiological functions necessary for survival and fertility. The average life-span of HGPS patients is 13 years. To date, no curative treatments for HGPS are available. In 2003, mutation in the gene Lamin A was found to be responsible for HGPS. Why and how malfunctioning Lamin A could cause premature aging was however still not known.
The Hutchison-Gilford Progeria Syndrome (HGPS) is a disease that causes the onset of severe premature aging. HGPS patients have elderly-like appearance and rapid deterioration of physiological functions necessary for survival and fertility. The average life-span of HGPS patients is 13 years. To date, no curative treatments for HGPS are available. In 2003, mutation in the gene Lamin A was found to be responsible for HGPS. Why and how malfunctioning Lamin A could cause premature aging was however still not known.
A research team led by Dr Zhong-jun Zhou has created a mouse model closely resembling the human disorder of HGPS. By studying this mouse model and cells from HGPS patients, they found that malfunction of Lamin A impairs the repair of DNA damages and the resulting genomic instability contributes to premature aging. Their findings have helped us understand the normal aging process and to develop novel anti-aging strategies.
The Use of Zebrafish Model to Study Gene Functions
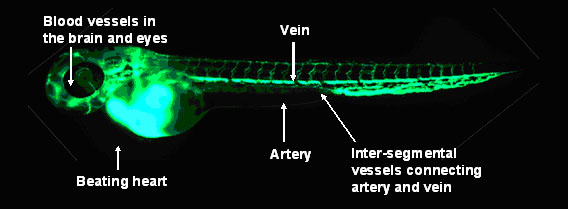 Genetically engineered Zebrafish with circulating red blood cells and blood vessels exhibiting green fluorescence.
Genetically engineered Zebrafish with circulating red blood cells and blood vessels exhibiting green fluorescence.
In collaboration with other laboratories in the United States and Mainland China, Dr Anskar Leung and his team have maintained colonies of genetically engineered zebrafish, in which the circulating red blood cells and blood vessels exhibit green fluorescence. Specific gene functions can be suppressed or over-expressed and their effects directly visualized real-time in live whole-organisms. With these cutting-edge technologies, they can interrogate the roles of specific genes in the regulation of important processes during normal and disease (such as cancer) development.